Alpha-Cell Dysfunction Should Be
the Focus of the New T1D Model
Let’s start the search for a new model by shifting the focus from the beta-cells to the alpha-cells. Some experts have proposed that alpha-cell dysfunction in T1D outweighs the failure to correctly mimic endogenous insulin secretions.
Alpha-cells play a central role in maintaining euglycemia, because glucagon is the primary control mechanism for regulating endogenous glucose influx from the liver to the blood stream. Liver output of glucose is directly proportional to plasma glucagon levels. See reference [1] for an overview of current views (2017) of the roles of insulin and glucagon in glucose homeostasis.
In addition to the canonical role of glucagon in glucose homeostasis, there is long established evidence that glucagon plays a role in energy homeostasis by enhancing satiety, increasing energy expenditures, and inducing thermogenesis. [2]
Alpha-cells are known to respond to nonglycemic plasma signals, e.g. arginine: [3]
“Amino acid-stimulated glucagon secretion during meals has a different purpose (from protecting against hypoglycemia): amino acids stimulate insulin secretion, which mobilizes amino acid transporters and effects their storage in peripheral tissues. At the same time, insulin obligatorily recruits GLUT-4 glucose transporters in muscle and fat. The hypoglycemic potential of such GLUT4 mobilization is averted only by the simultaneous liberation of endogenous glucose in response to feedforward (anticipatory) glucagon secretion.” [4]
In healthy, nondiabetic subjects, alpha-cell secretion of glucagon is responsive to the need to restore euglycemia in response to rising or falling blood glucose, and also to respond to nonglycemic signals:
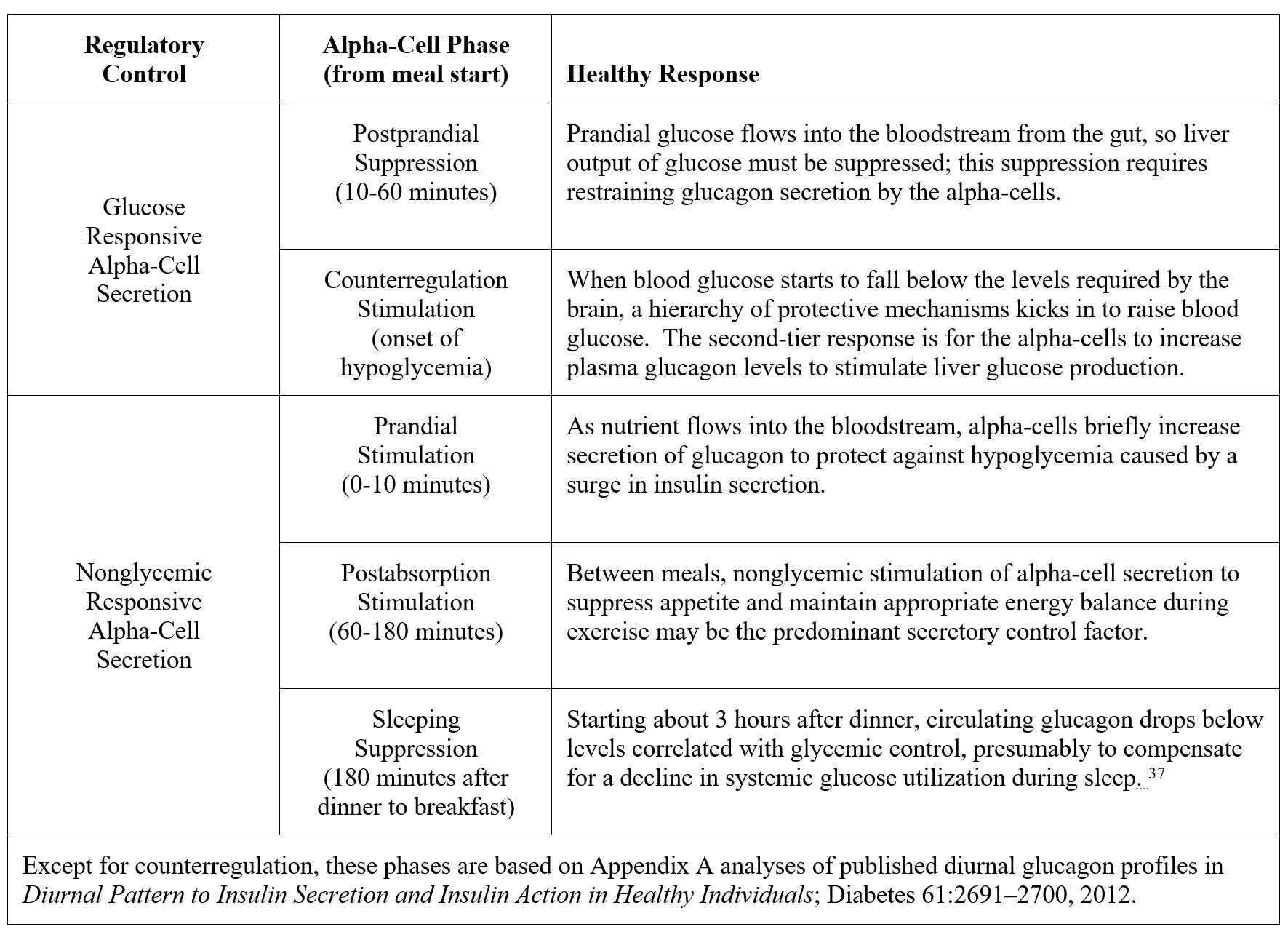
Two not mutually exclusive observations have promoted the idea that normalizing glucagon regulation could be the key to effective T1D therapy: excessive postprandial glucagon secretion encourages glucose influx from the liver thereby increasing the need for mealtime insulin boluses, and glucagon counterregulatory failure is the principal barrier to intensive insulin therapy aimed at normalizing blood glucose.
Suppressing postprandial
glucagon mitigates hyperglycemia
Unger et al have pointed out that T1D hyperglycemia is aggravated by excessive postprandial glucagon secretion: [5] “The present studies demonstrate that failure to suppress glucagon following glucose ingestion exacerbates postprandial hyperglycemia in T1D subjects. These data indicate that therapy for T1D subjects is unlikely to result in completely normal carbohydrate tolerance unless both the concentration and pattern of change of glucagon following glucose ingestion is also restored to normal.” [6]
Concern about this “Prandial Problem” of hyperglycemia has led to the suggestion that diabetes drug R&D shift from basal to prandial therapy: “The prandial problem, including postprandial hyperglycemia, weight gain, and hypoglycemia caused by overreliance on injected insulin, is an endocrine and neurologic puzzle that calls for further basic and clinical research. (2017)” [7]
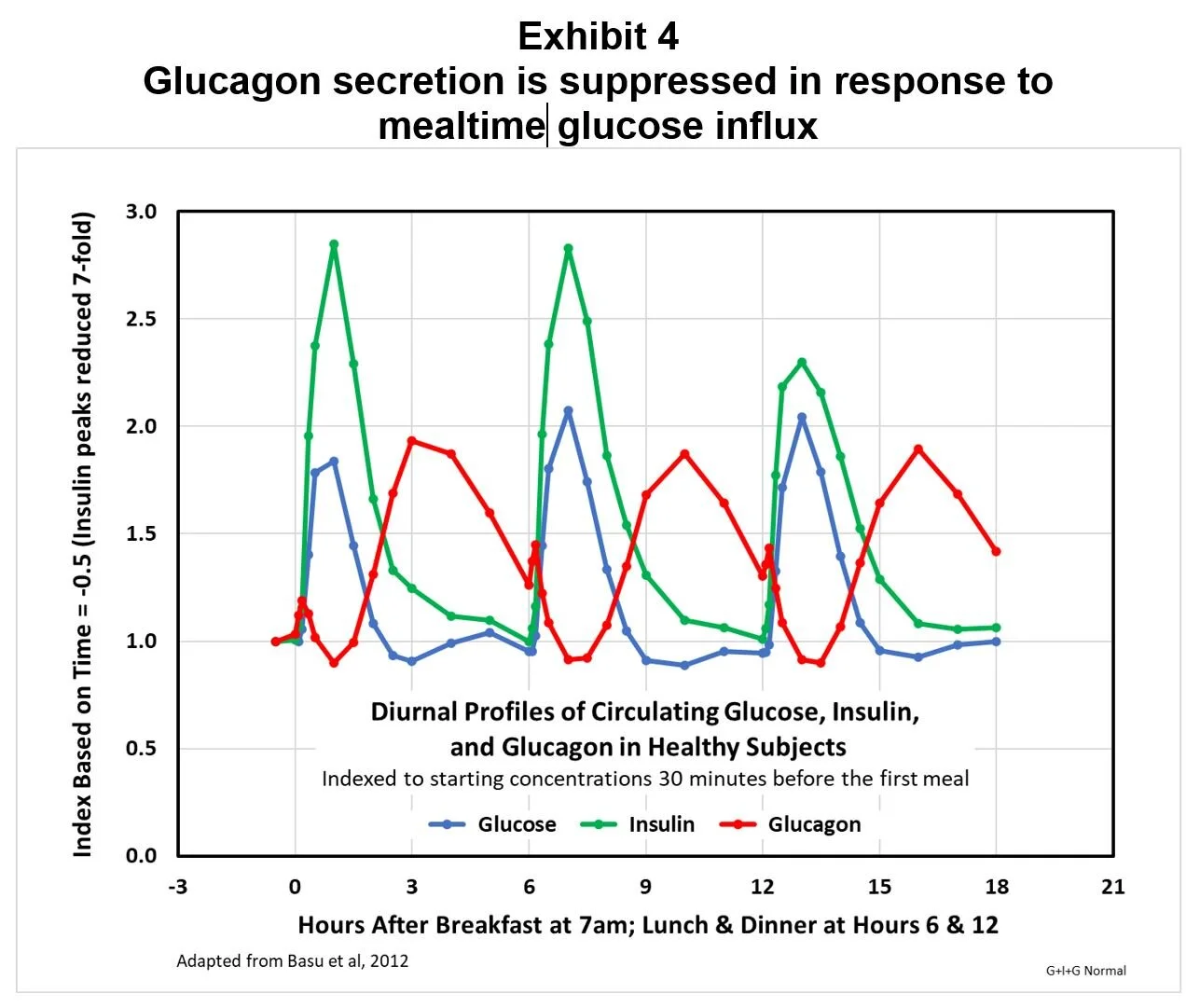
In nondiabetics, following a momentary, arginine-induced spike, prandial increases in circulating glucose cause immediate reductions in circulating glucagon. Exhibit 4 demonstrates this by showing for healthy, nondiabetic subjects glucose, insulin, and glucagon diurnal profiles indexed to preprandial basal levels, with the insulin index divided by seven to allow visual comparisons; in this study, following the amino-acid induced surge, postprandial circulating glucagon was suppressed to a level about 10% below basal levels.
In healthy, nondiabetic subjects, circulating insulin levels are tightly correlated to circulating glucose levels, because beta-cell secretion is mostly responsive to glucose. Linear regression analysis of three different studies confirms that 90%+ of insulin diurnal profiles are explained by glucose profiles:

In contrast, circulating glucagon is much less closely correlated with circulating glucose following ingestion of simple carbohydrate, and not at all correlated to glucose following complex carbohydrate and in T1D:
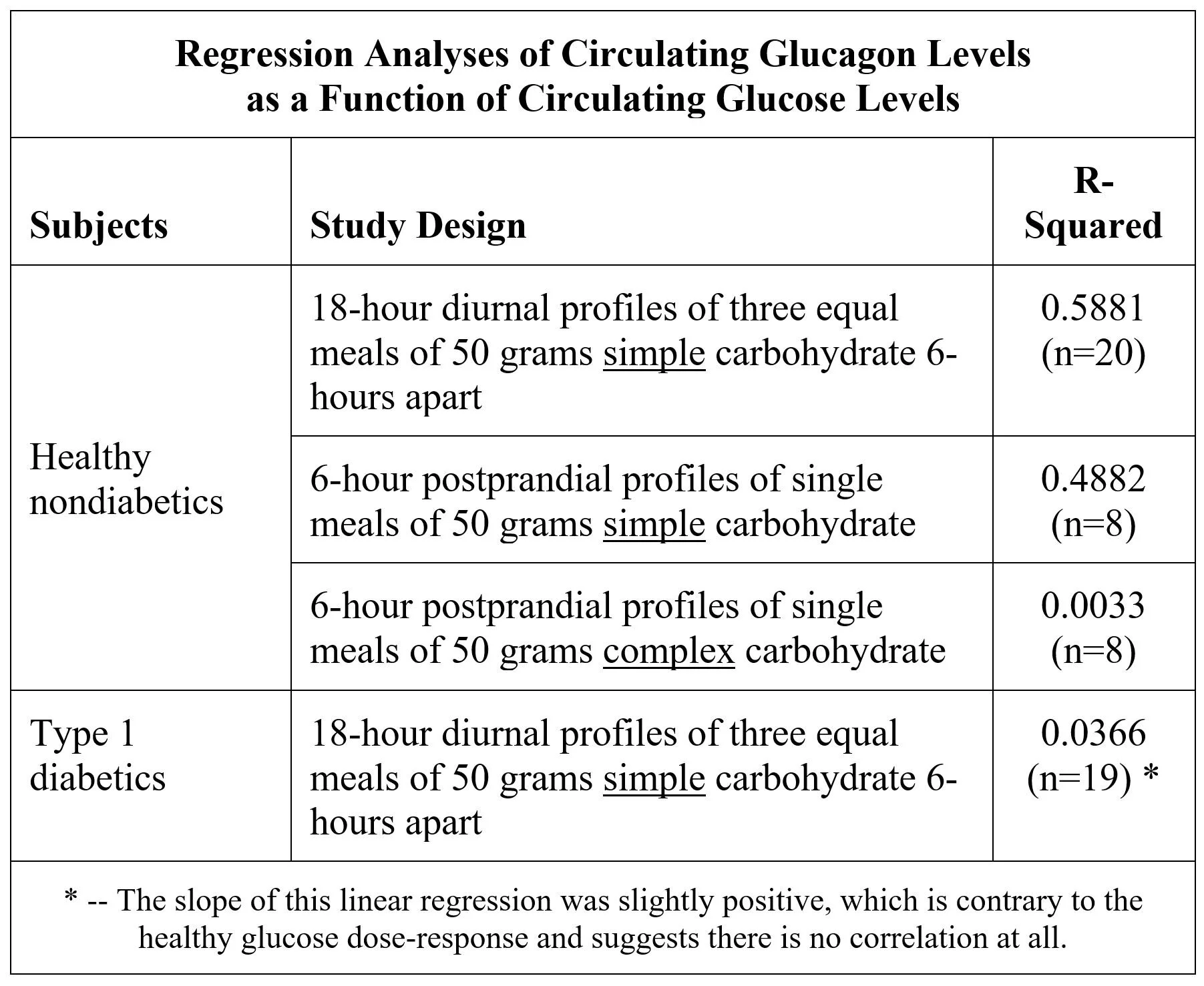
Exhibit 5 compares the glucagon dose-response to changes in glucose from a simple carbohydrate meal for nondiabetic and T1D subjects in the diurnal studies.
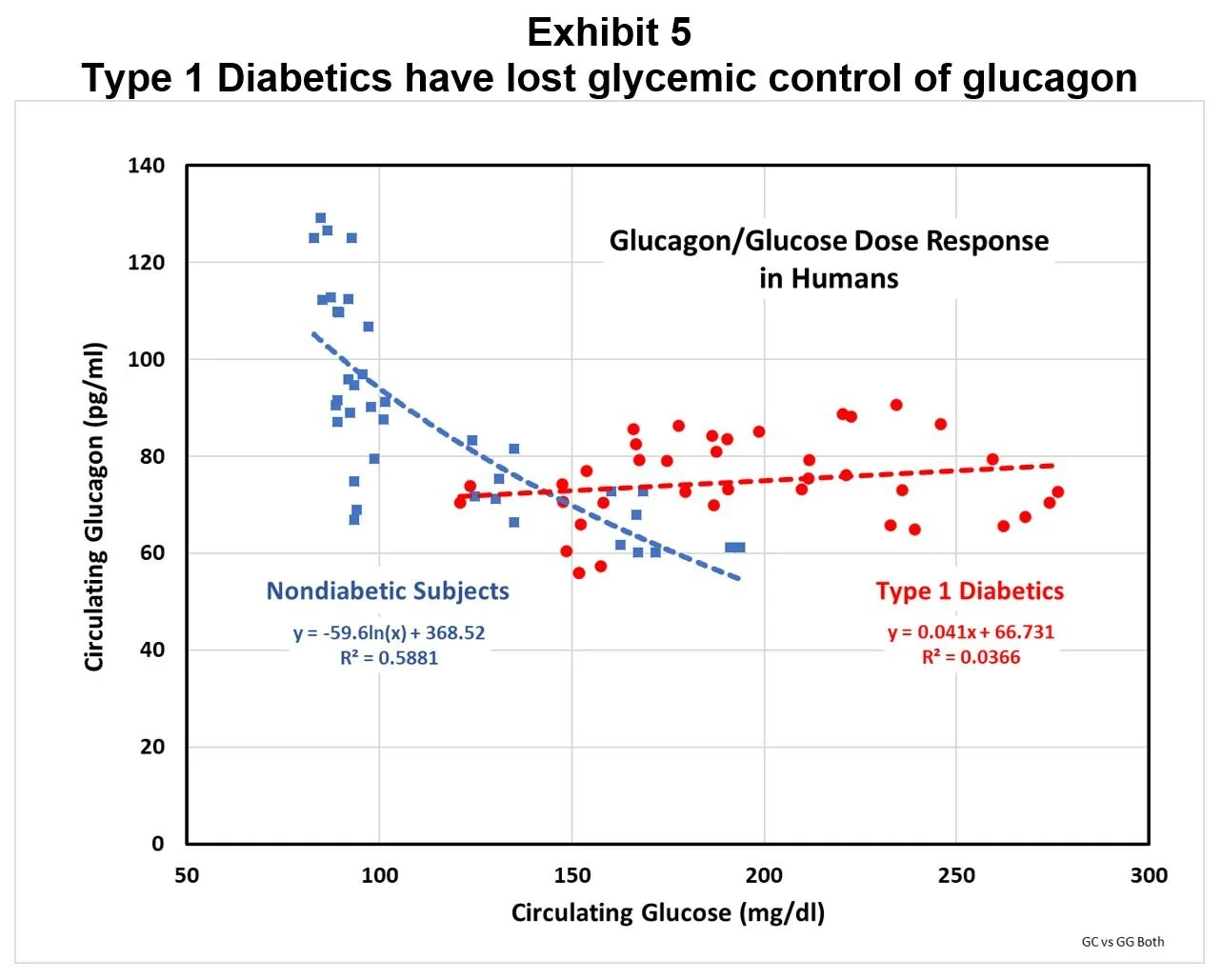
It’s clear from Exhibit 5 that T1D subjects have circulating glucagon levels mostly well above the levels predicted by their chronic hyperglycemia, based on the nondiabetic dose-response. If the T1D diurnal glucose profiles are used in the nondiabetic correlation equation to predict circulating glucagon, the resulting area under the curve compared to actual T1D levels indicates that T1D subjects are exposed to 35% more circulating glucagon than they should be, i.e. they are relatively hyperglucagonemic.
Since glucagon stimulates hepatic glucose production, an excess of glucagon can be expected to contribute to the chronic hyperglycemia of T1D. This is supported by a T1D animal experiment in which correcting the excessive glucagon secretion virtually eliminated the need for insulin. [8] In this experiment plasma glucose was normalized in diabetic rats given leptin to suppress their secretion of glucagon; results are shown in Exhibit 6.
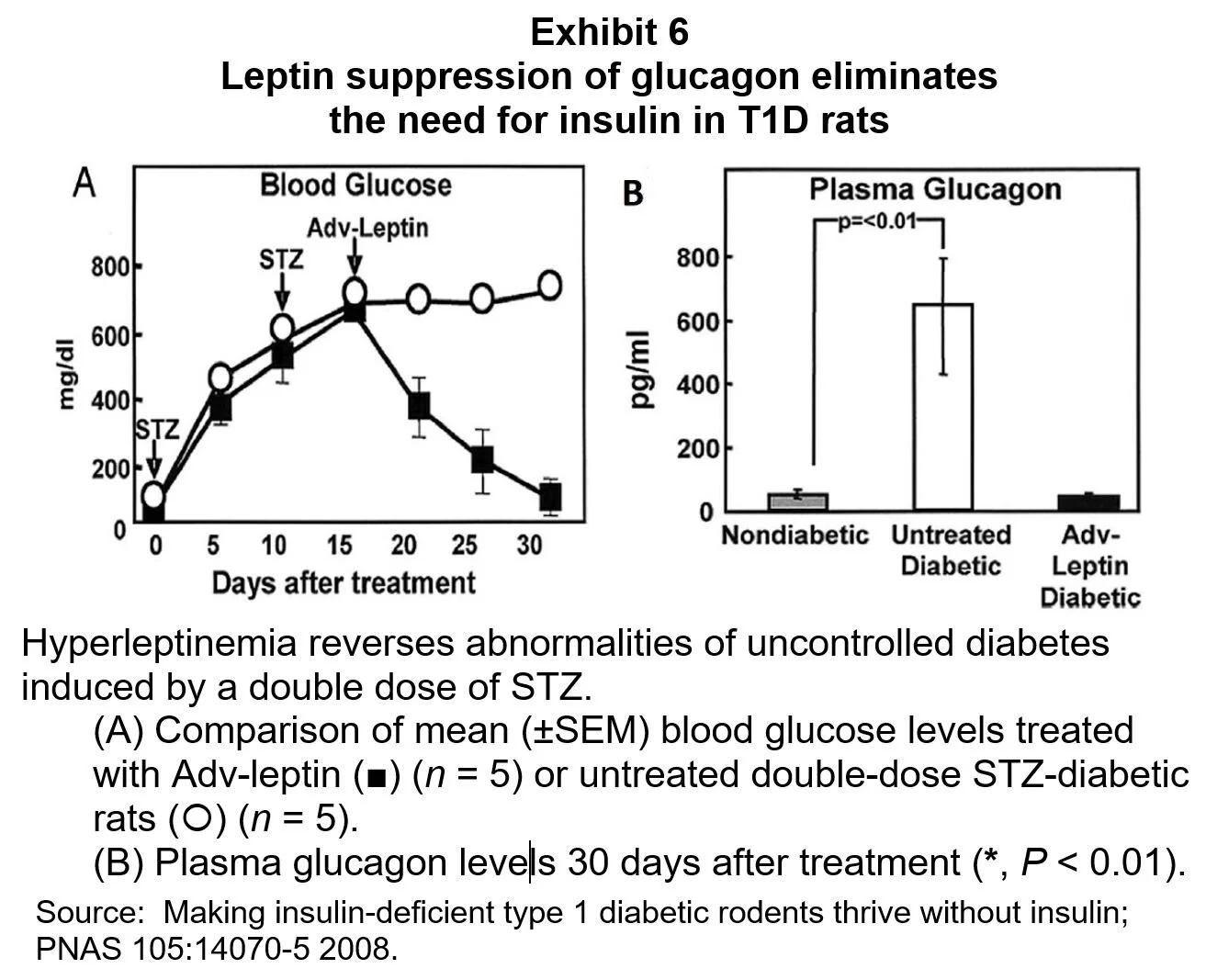
It seems clear that dysregulated alpha-cell secretion is a key player in postprandial hyperglycemia.
Counterregulatory failure
is a treatment barrier
Cryer et al point out that the risk of hypoglycemia is the biggest barrier to normalizing glucose in T1D, as described in a June 2018 review: [9]
“Iatrogenic hypoglycemia is a fact of life for most people with T1D who must, of course, be treated with insulin. Most have untold numbers of episodes of asymptomatic hypoglycemia which are not benign since they impair defenses against subsequent hypoglycemia. They suffer an average of two episodes of symptomatic hypoglycemia per week – thousands of such episodes over a lifetime of diabetes – and of about one episode of severe, at least temporarily disabling, hypoglycemia per year. Hypoglycemia causes brain fuel deprivation that, if unchecked, results in functional brain failure that is typically corrected after the plasma glucose concentration is raised. Rarely, if it is profound and prolonged, can result in brain death. Hypoglycemia may lead to cardiac arrhythmias, especially in patients with preexisting cardiac abnormalities. Additionally, hypoglycemia has been demonstrated to be pro-coagulant and pro atherothrombotic. Furthermore, severe hypoglycemia has been associated with increased risk of death extending many months after the sentinel episode. Early reports suggested that 2 to 4% of deaths of people with diabetes, largely T1D, were the result of hypoglycemia. More recent reports suggest that 6 to 10% of deaths of people with T1D are the result of hypoglycemia. Regardless of the actual rate, the fact that there is an iatrogenic hypoglycemia mortality rate is alarming.”
The risk of serious hypoglycemia is inversely correlated with the intensity of insulin therapy aimed at lowering HbA1c. [10] As insulin therapy is intensified to achieve lower average blood glucose, the risk of serious hypoglycemia increases, with the result that patients are inclined to live with a hyperglycemia “buffer.”
Hypoglycemia is a risk because glucagon counterregulation is defective in T1D, i.e. the alpha-cells in T1D do not respond to hypoglycemia by increasing glucagon secretion. [11] “This is a signaling defect; glucagon secretory responses to stimuli other than hypoglycemia are largely, if not entirely, intact. The mechanism of the absent glucagon response to hypoglycemia that characterizes established type 1 diabetes is not known, but it is linked tightly to, and is possibly the result of, endogenous insulin deficiency.” [12]
* * * * * * *
The unifying conclusion between the Unger and Cryer camps is that T1D therapy would be more effective if drug therapy could restore glycemic regulation of alpha-cell secretion:
Appropriate postprandial glucagon suppression would reduce the glycemic burden of hepatic glucose production so that less insulin would be needed at mealtimes, thus lowering the risk of iatrogenic hypoglycemia.
Restoration of the glucagon counterregulatory response would mitigate the risk of hypoglycemia and permit relatively more aggressive insulin therapy.
We agree with those who believe targeting alpha-cell secretion is also a key to reducing blood glucose variability, which is increasingly viewed as a primary cause of diabetic complications. [13]
Note: In this discussion of alpha-cell regulation we refer to analyses of diurnal and postprandial glucagon profiles applied to data generated by Andy Basu’s team at the Mayo Clinic as presented in Appendix A:
Diurnal pattern to insulin secretion and insulin action in healthy individuals; Diabetes 61:2691-700 2012.
Diurnal pattern of insulin action in type 1 diabetes; Diabetes 62:2223-9 2013.
A novel natural tracer method to measure complex carbohydrate metabolism: Am J Physiol Endocrinol Metab 317:E483-93 2019.
Endnotes:
[1] Insulin and glucagon: partners for life; Endocrinology 158(4):696-701 2017.
[2] -- Glucagon Control on Food Intake and Energy Balance; International Journal of Molecular Sciences 20:3904-16 2019.
[3] -- Arginine-Stimulated Acute Phase of Insulin and Glucagon Secretion in Diabetic Subjects; Journal of Clinical Investigation 58:565-70 1976.
[4] Inhibition of glucagon secretion; chapter in Amylin: Physiology and Pharmacology, page 151 2005.
[5] Glucagon is the key factor in the development of diabetes; Diabetologia 59:1372-5 2016.
[6] Failure of glucagon suppression contributes to postprandial hyperglycaemia in IDDM; Diabetologia 38:337-43 1995.
[7] Basal glucose can be controlled, but the prandial problem persists – it’s the next target!; Diabetes Care 40-291-300 2017.
[8] Making insulin-deficient type 1 diabetic rodents thrive without insulin; PNAS 105:14070-5 2008.
[9] Hypoglycemia during therapy of diabetes; Endotex at www.endotex.org June 2018.
[10] Frequency and Morbidity of Severe Hypoglycaemia in Insulin-treated Diabetic Patients; Diabetic Medicine 10:238-45 1993.
[11] Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha-cell defect; Science 182: 171-3 1973.
[12] Hypoglycemia in diabetes; Diabetes Care 26:1902-12 2003.
[13] Glycemic variability and diabetes complications: Does it matter? Of course it does!; Diabetes Care 38:1610-14 2015.