The New Model Focuses on
Alpha-Cell Sensing of Hypoglycemia
Let’s start with a controversial puzzle: what disrupts the glucagon counterregulatory response in T1D. “While the molecular mechanisms involved in the regulation of insulin secretion are well understood, knowledge of those that mediate the inhibition of glucagon release remains fragmentary.” (December 2018 alpha-cell review.) [1]
Glucagon secretory dysfunction in T1D is probably not due to a global defect in alpha-cell function. They remain normal in number and histological appearance in T1D, and the alpha-cell response to ingested arginine is normal or increased in T1D. [2]
As shown in Exhibit 7, while the amplitude of the T1D diurnal glucagon profile is muted compared to the nondiabetic profile, the T1D pattern is similar.
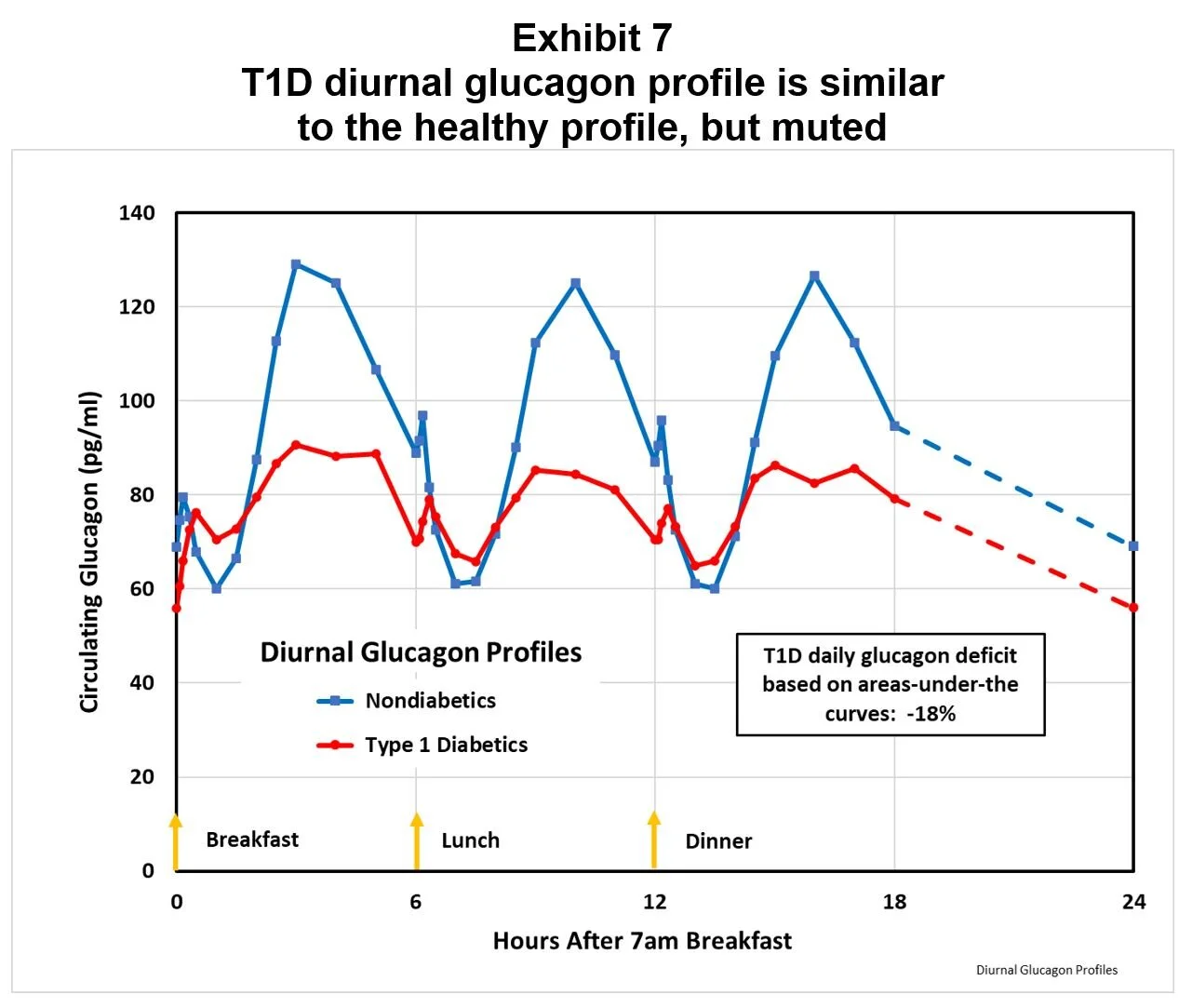
This similarity of diurnal profiles suggests that the nonglycemic regulation of alpha-cells remains intact, a finding confirmed in studies dating back to 1973. 44
Hypoglycemia sensing appears to lie at the root of defective alpha-cell response, and existing theories of alpha-cell hypoglycemia sensing can be classified into three not mutually exclusive mechanisms:
DIRECT signaling of plasma glucose on alpha-cells: Isolated alpha-cells have been shown in some experiments to respond to blood glucose levels, but the direct response to hypoglycemia appears to be relatively weak. [3] At the RNA level, glucose regulates proinsulin and prosomatostatin, but not proglucagon. [4] If this were the only, or even most important, glucose sensing mechanism, grossly defective glucagon counterregulation would not be expected to characterize T1D, because the alpha-cells remain sensitive to non-glycemic signals in T1D.
PARACRINE signaling of beta-cell secretions to alpha-cells: Some data (Cryer et al) suggest alpha-cells respond to beta-cell secretions. Over time, T1D patients’ decline in C-peptide is mirrored by increasing postprandial glucagon secretions. 69 Insulin, or perhaps zinc, is assumed to be the paracrine messenger. [5] [6] This paracrine hypothesis is the basis for the “insulin switch-off model:” insulin secretion downregulates glucagon secretion, especially at mealtime, and hypoglycemia suppresses insulin secretion, causing a glucagon secretion rebound. However, some data (Unger et al 1983) dispute this idea: “These results indicate that the glucagon response to insulin-induced hypoglycemia is independent of the level of both endogenous intraislet and exogenous arterial insulin concentration in normal man, and that this response may be normal in the absence of endogenous insulin secretion, in contrast to earlier reports. Thus, loss of beta cell function is not responsible for alpha cell failure during insulin-induced hypoglycemia in IDDM.” [7] Suffice it to say that the data with respect to intra-islet regulation of glucagon secretion is contradictory and confusing, probably because it does not consider the possible role of a potent alpha-cell suppressor, the neuroendocrine hormone amylin.
CENTRAL NERVOUS SYSTEM signaling of hypoglycemia to alpha-cells: Autonomic activation is an important player in glucose counterregulation, [8] and the concept of brain control of glucose homeostasis dates back over 150 years to work by Claude Bernard. [9] But a focus on pancreatic regulation by endocrine hormones has overshadowed research on the CNS glucosensory network. [10] The first response to a dangerous fall in glucose is detection by hypoglycemia sensors, including neurons in the hypothalamus and other regions. [11] [12] [13] “This indicated that the brainstem may be the most physiologically relevant site of hypoglycemia detection and counterregulation.” [14]
We favor the view that the CNS is the main mediator of alpha-cell response to hypoglycemia. Substantial evidence exists that autonomic nerves are of major importance for the glucagon response to hypoglycemia in healthy humans and experimental animals. [15] Ganglionic blockade eliminates 75-90% of glucagon counterregulation without affecting glucagon response to arginine administration. [16] Data indicate that the magnitude of the glucagon response to iatrogenic insulin is dependent on the recognition of hypoglycemia by the brain, not the islet. [17]
An experiment in dogs demonstrated that counterregulation is primarily triggered in the brain, not in the pancreas. When dogs were made hypoglycemic, the alpha-cells responded strongly; but, when glucose was infused directly into the brain to maintain local euglycemia, counterregulation was blocked (Exhibit 8).
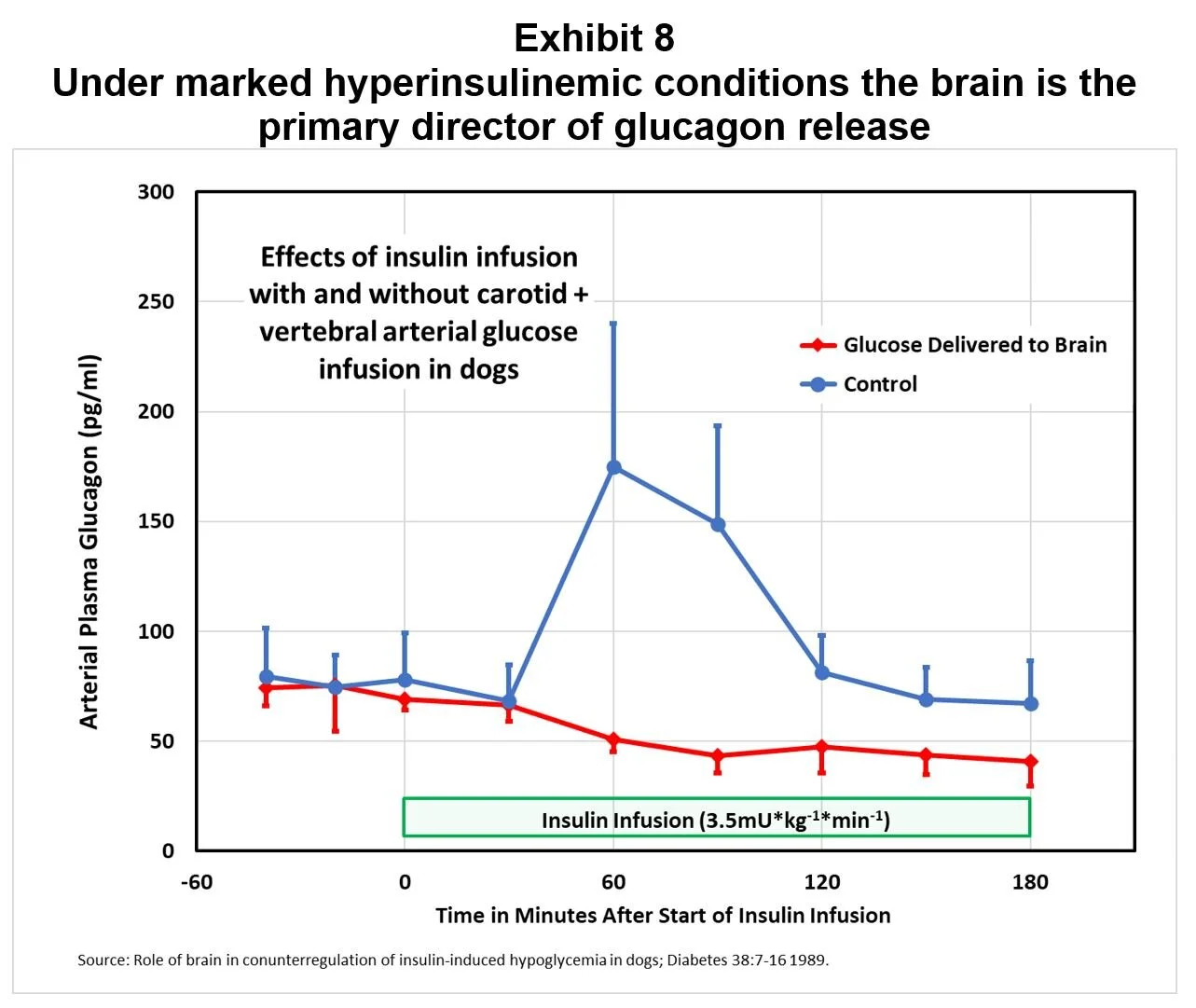
“These results suggest that under marked hyperinsulinemic conditions the brain is the primary director of glucagon release and that it is responsible for ~75% of the life-sustaining glucose production.” [18]
CNS control makes teleological sense. Although the adult human brain constitutes only ~2% of body weight, it accounts for ~20% of whole-body glucose utilization. [19] The brain uses 60-80% of blood glucose in a resting state, and the brain cannot store more than a 20-minute supply of glycogen, with the result that low plasma glucose concentrations quickly cause functional brain failure. [20] Thus, the tissue most vulnerable to hypoglycemia is logically the tissue where hypoglycemia is detected.
In summary, we believe that the beta-cells are the primary regulator of alpha-cell response to hyperglycemia, while the brain is the primary regulator of alpha-cell response to hypoglycemia. And we propose that amylin is the signaling mechanism which activates both functions. We now show how a simple systems analysis can support this hypothesis.
Endnotes:
[1] The alpha cell in diabetes mellitus; Nature Reviews Endocrinology; 14:694-704 2018.
[2] To much glucagon, too little insulin; Diabetes Care 31:1403-4 2008.
[3] Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells; Diabetologia 50:370-9 207.
[4] Glucose regulates proinsulin an prosomatostatin but not proglucagon messenger ribonucleic acid levels in rat pancreatic islets; Endocrinology 141:174-80 2000.
[5] Loss of the decrement in intraislet insulin plausibly explains loss of the glucagon response to hypoglycemia in insulin-deficient diabetes – documentation of the intraislet insulin hypothesis in humans; Diabetes 54:757-64 2005.
[6] Zinc, not insulin, regulates the rat alpha-cell response to hypoglycemia in vivo; Diabetes 56:1107-12 2007.
[7] Mechanisms of glucagon secretion during insulin-induced hypoglycemia in man – Role of the beta-cell and arterial hyperinsulinemia; Journal of Clinical Investigation 73:917-22 1984.
[8] Minireview: The role of the autonomic nervous system in mediating the glucagon response to hypoglycemia; Endocrinology 153(3):1055-62 2012.
[9] Peripheral and central glucose sensing in hypoglycemic detection; Physiology 29(5):314-24 2014.
[10] The new biology and pharmacology of glucagon; Physiol Rev 97:721-66 2017.
[11] Central mechanisms of glucose sensing and counterregulation in defense of hypoglycemia; Endocrine Reviews published online January 22 2019.
[12] Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release; Diabetes 44:180-4 1995.
[13] ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis; Nature Neuroscience 4:507-12 2001.
[14] Brain glucose sensing and neural regulation of insulin and glucagon secretion; Diabetes, Obesity and Metabolism 13(Suppl. 1):82-8 2011.
[15] Autonomic regulation of islet hormone secretion – implications for health and disease; Diabetologia 43:393-410 2000.
[16] Activation of autonomic nerves and the adrenal medulla contributes to increased glucagon secretion during moderate insulin-induced hypoglycemia in women; Diabetes 46(5):801-807 1997.
[17] The physiology of glucagon; Journal of Diabetes Science and Technology 4:1338-44 2010.
[18] Role of brain in counterregulation of insulin-induced hypoglycemia in dogs; Diabetes 38:7-16 1989.
[19] Hypoglycemia in Diabetes: Pathophysiology, Prevalence, and Prevention; book by Philip Cryer 2016.
[20] Glucagon and the cell – Physiology and pathophysiology part 1; NEJM 304:1518-24 1981.