Systems Analysis Supports the
Idea of an Alpha-Cell Inhibitor
It’s been proposed that distributed glucose sensors provide critical inputs to an integrative network, allowing for more finely tuned responses to glycemic challenges. Glucagon, like insulin, is secreted in a basal and pulsatile manner, and studies show pulsatile delivery of glucagon is more potent than continuous delivery, probably because of receptor dynamics. Continuous delivery may suppress receptor expression, or receptors may be more responsive to rate of change in concentration; the latter is supported by the observation that there is a glucagon dose-response to simple carbohydrates that rapidly increase circulating glucose, but there is no dose-response to complex carbohydrates which enter circulation at about half that rate (Appendix A). Alpha-cell pulses are produced in apparently linked antiphasic manner to those of insulin and somatostatin, which appear to be highly regulated through a series of controlling influences from within and without the pancreas.
These observations are consistent with the idea that alpha-cells are under constant tonic inhibition by the beta-cells as postulated by the insulin switch-off model. Tonic inhibition of glucagon in T1D rodents reduces glycemic volatility consistent with the insulin switch-off model. [1] However, in a network system, looking at individual interactions doesn’t provide a good picture of what’s going on, as demonstrated by the apparently contradictory data about alpha-cell sensing. What’s needed is a system-level approach combining in vivo and in silico studies.
To this end, an in-silico systems analysis is a new, more comprehensive way of analyzing the switch-off model of glucagon mediated counterregulation. Farhy et al have demonstrated how counterregulation abnormalities can be simulated with a minimal systems network. [2]
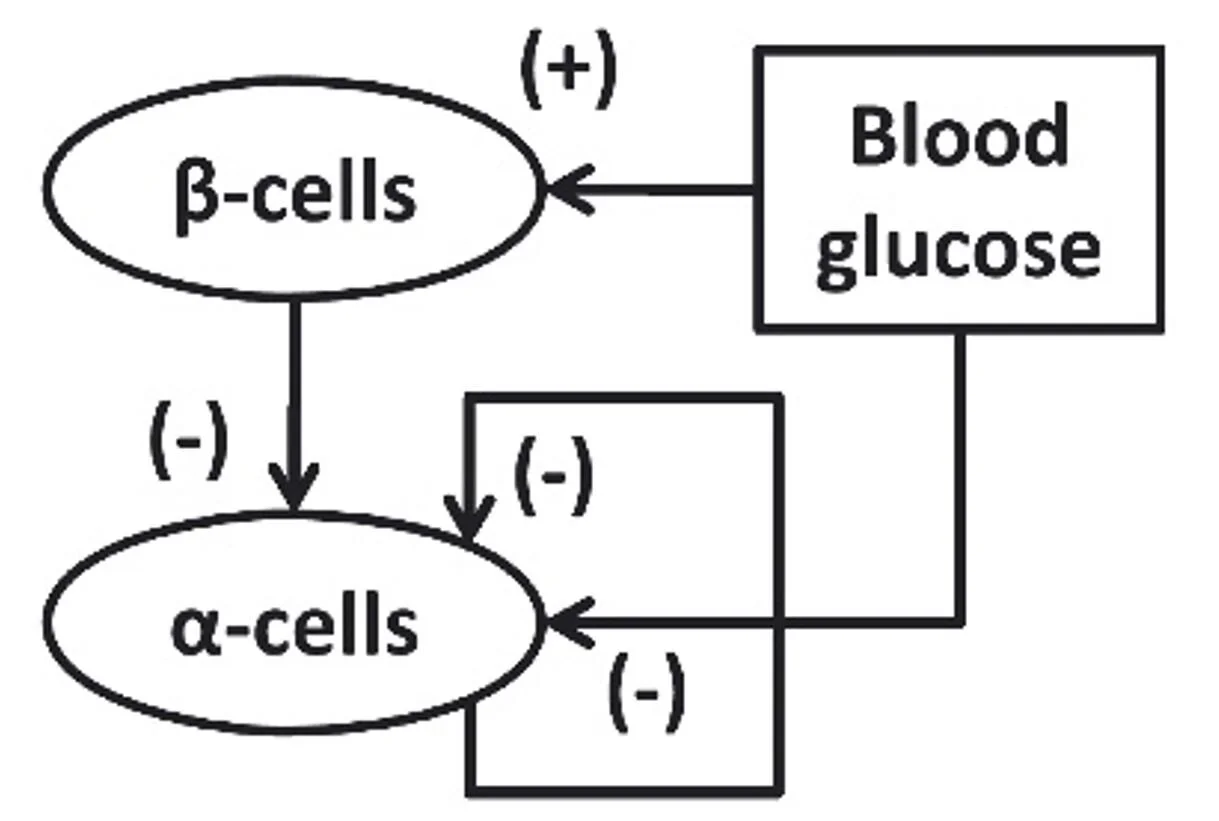
Their model builds upon the premise that the specific trigger is probably from the beta-cell. They consider the counterregulatory defect as the failure of a dynamic minimal control network to respond adequately to a hypoglycemia stimulus, and they interpret glucagon counterregulation as a rebound in response to switch-off of the putative alpha-cell inhibitor signal.
Their counterregulatory model simulates glucagon control at this simple systems level:
In healthy euglycemia, beta-cells generate an alpha-cell inhibitor signal which restrains glucagon secretion. When hypoglycemia occurs, beta-cells shut down secretion of the alpha-cell inhibitor, and alpha-cells rebound with robust glucagon secretion.
T1D removes the alpha-cell inhibitor signal from the beta-cells, thereby eliminating hyperglycemic control of glucagon secretion, as well as eliminating the tonic inhibition of glucagon secretion required for the shut-off rebound response to hypoglycemia.
As an intervention in T1D, the modelers used somatostatin in animals to validate their in silico model. [3] But somatostatin is not a practical solution given the potential side effects of somatostatin and the requirement to quickly lower plasma levels at the onset of hypoglycemia.
As discussed above leptin has also been proposed as an alpha-cell inhibitor in T1D, because leptin’s glucose lowering effects are accompanied by normalization of plasma glucagon levels. [4] Preclinical results were encouraging; however, a human pilot study was disappointing.
Thus, we believe the search for a pharmaceutical solution to restoring counterregulation in T1D should focus on finding a drug candidate which satisfies the following criteria:

We propose that an amylin agonist is probably that inhibitor.
Endnotes:
[1] Paracrinology of islets and the paracrinopathy of diabetes; PNAS 107:16009-16012 2010.
[2] Optimizing reduction in basal hyperglucagonaemia to repair defective glucagon counterregulation in insulin deficiency; Diabetes, Obesity and Metabolism 13:133-43 2011.
[3] Amplification of pulsatile glucagon counterregulation by switch-off of alpha-cell-suppressing signals in streptozotocin-treated rats; Am J Physiol Endocrinol Metab 295:E575-85 2008.
[4] The role of leptin in diabetes: metabolic effects; Diabetologia 59:928-32 2016.